How are the scores computed?
The combined score is computed by combining the probabilities from the different evidence channels and corrected for the probability of randomly observing an interaction. For a more detailed description please see von Mering, et al. Nucleic Acids Res. 2005
To combine the scores we add the probabilities for each of the channels. To each channel, a 'prior' has been added to account for the probability that two randomly picked proteins are interacting. Before combing the channels the 'prior' has to be removed and then added back again to the combined score. Here is how the combined score is computed for an interaction.
-
For each of the scores for the individual channels (s_i) remove the prior (p=0.041):
s_i_noprior = (s_i - p) / (1 - p) -
Combine the scores of the channels:
s_running_total = 1 - s_running_total * (1 - s_i_noprior) -
Add the prior back (once):
s_total = s_running_total + p * (1 - s_running_total)
In code it would be something like this:
s_exp = 0.621
s_txt = 0.585
p = 0.041
s_exp_nop = (s_exp - p) / (1 - p)
s_txt_nop = (s_txt - p) / (1 - p)
s_tot_nop = 1 - (1 - s_exp_nop) * (1 - s_txt_nop)
s_tot = s_tot_nop + p * (1 - s_tot_nop)
Also, homology correction is applied to the co-occurrence and text-mining scores.
effective co-occurrence score = co-occurrence score * (1 - homology score)
effective text-mining score = text-mining score * (1 - homology score)
If you would like to see the exact algorithm or calculate the combined score yourself you can utilize and modify the follwing python script.
What are local STRING network clusters? Top ↑
Local STRING network clusters or simply STRING clusters are precomputed protein clusters derived by hierarchically clustering the full STRING network using an average linkage algorithm. The smallest clusters generated consist of 5 proteins and largest used in the enrichment analysis contain 200 proteins. In order to reduce redundancy we remove all clusters with a size difference of less than 5 proteins towards their child cluster (next smallest cluster in the hierarchy). The names are derived automatically based on a cluster’s consensus protein annotations taken from GO, KEGG, Reactome, UniProt, Pfam, SMART, and InterPro. All clusters and their hierarchical tree are available for download in the download section of STRING
How can I obtain the complete data set? Top ↑
STRING is completely free and avaliable to download for both, our academic and commercial users. In the download section of STRING you can find all the flat-files that will let you generate your own STRING network. Most of the other data STRING uses to generate the evidences behind the links can be found in the PostgreSQL dump of STRING database. Unfortunetly due to licencing issues we had to remove KEGG data and textmining snippets from these SQL tables.
How do I change the color of the nodes? Top ↑
There are several methods avaliable to label your STRING network with differently colored nodes.
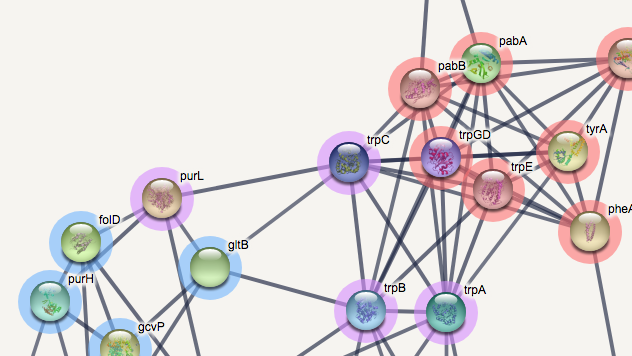
- Payload mechanism. While you won't be able to recolor the nodes themselves the STRING's payload mechanism allows you to add a halo in a specified color around the STRING nodes. In order to upload your payload data, go to "My Data" (found in the upper right corner). When in "My Data", below your search history, you can find "add new payload dataset". Click it and follow the instructions. You can click on the given examples to see the format of the payload (the second column specifies the color in HEX values). After you successfully uploaded your payload it shoud show up under "your payload datasets". Click show->Show Nodes In Network to view the colored network. It should look something like this:
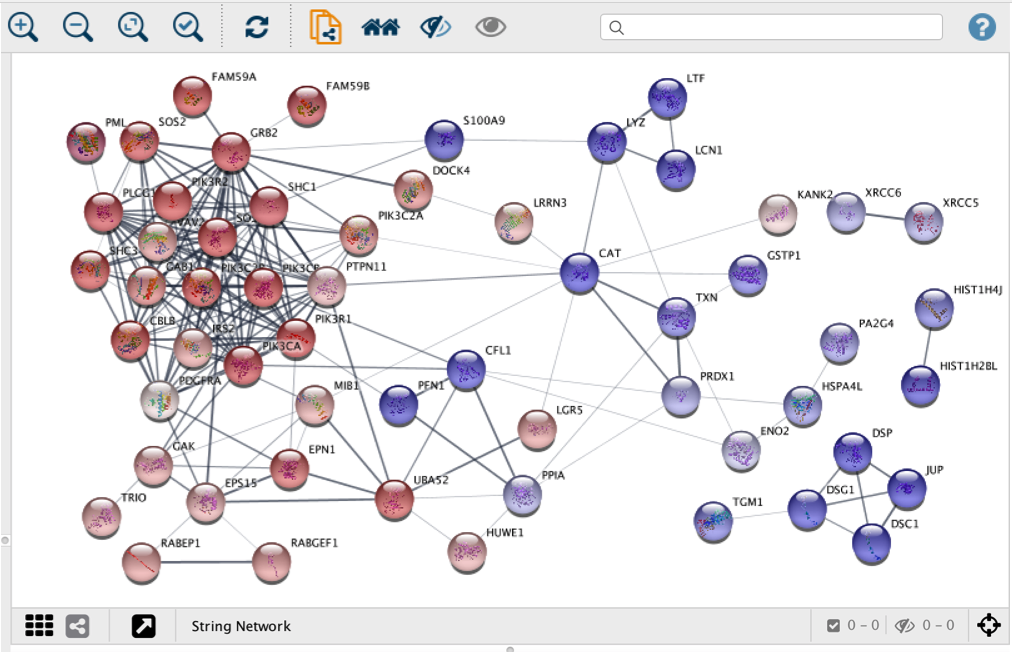
- Cytoscape stringApp. Our new Cytoscape app preserves the look of STRING network but retains the powerful tools Cytoscape is known for. These include, among many others, recoloring of the nodes based on the given experimental values, network clustering and the diverse layout functionality.
For more information please see our stringApp webpage
- Python script. The script allows you to assign arbitrary colors to protein nodes directly on the network created through the STRING website. Go to the "Export" tab (found below the network) and download the network as an SVG (vector graphics). Download the python script here. Open the Terminal and run the script on the SVG file like this: [NOTE: windows users need to install python first]:
python change_STRING_colors.py -s string_vector_graphic.svg
this will create a color_table.tsv (tab separated values) file, which you can open and modify in Excel, Google Spreadsheet, or any simple text editor (like NotePad, but not Word). The file should look like this:
| Node | color |
|---|---|
| pabC | rgb(255,255,255) |
| trpE | rgb(14,178,0) |
| trpGD | rgb(187,255,101) |
| trpC | rgb(178,171,0) |
| trpA | rgb(255,0,0) |
| ... | ... |
Just modify the second column with a color you would like to give to a node (when you remove the node from this file the node in the network will become white). For color values you can use standard english HTML color names, HEX color values, or RGB color values, all of which you can find here. After you assign the colors export the table as a TSV file (in Excel: File->Save As->Tab Delimited File). Now run the python script again, this time specifying your new color file, like this:
python change_STRING_colors.py -s string_vector_graphic.svg -c modified_colors_table.txt
This will generate new file with a suffix ".new_colors.svg". This file can be opened in your internet browser or any program which supports vector graphics where you can save it as BMP, PNG and JPG.
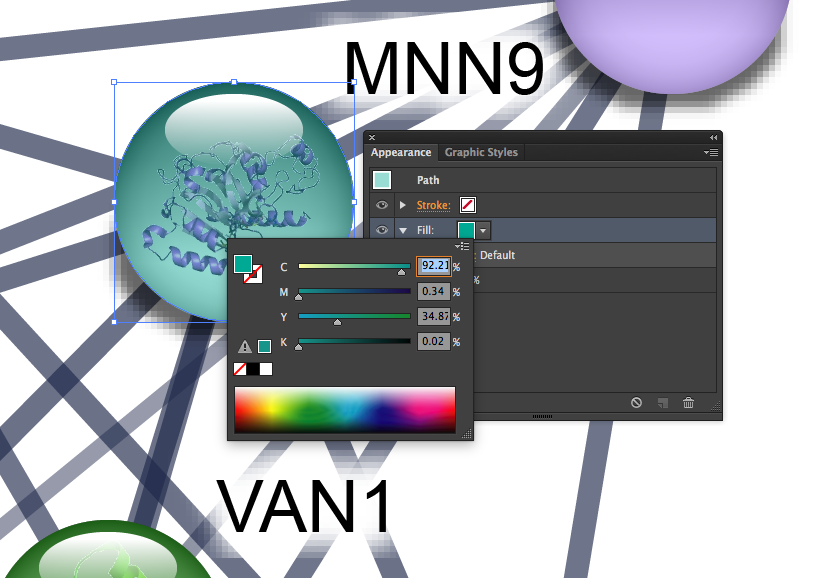
 4. Manually. If your network is not large and you just want to make small modifications the easiest way is to modify the color manually. Go to the "Export" tab (found below the network) and download the network as an SVG. You can open the file in a graphics program which supports vector graphics. When you see the network click on the node first to ungroup it. One of the layers of the node is the color circle, which can be manually modified. From there you can export the whole file as a BMP, PNG or JPG.
4. Manually. If your network is not large and you just want to make small modifications the easiest way is to modify the color manually. Go to the "Export" tab (found below the network) and download the network as an SVG. You can open the file in a graphics program which supports vector graphics. When you see the network click on the node first to ungroup it. One of the layers of the node is the color circle, which can be manually modified. From there you can export the whole file as a BMP, PNG or JPG.
I am interested in retrieving data of a few particular interaction for my script. How do I go about to get it? Top ↑
Please use the API If you plan to submit thousands of HTTP requests first make sure if the information you are seeking is not avaliable to download. If you still would like to use API please pause for at least a second between each API call. Too many concurent calls may slow down the server for all users.
How can I save a certain network? Top ↑
You can find your network avaliable for download under Tables/Exports tab. The network is avaliable to download in the variety of formasts: Bitmap Image, Scalable Vector Graphics, XML Summary (Proteomics Standards Initiative), Graph Layout, Protein sequences in FASTA format, and Text Summary of interaction scores.
For my latest manuscript, I would like to use a picture in svg-format produced by STRING. Must I ask for permission? Top ↑
Nope. But we appreciate if you cite us
How to cite STRING? Top ↑
Szklarczyk et al. Nucleic acids research 47.D1 (2018): D607-D613.2
How can I trace the origin of the different evidences for an interaction? Top ↑
This information is available if you click on an edge of the graph in the network view.
Which databases does STRING extract experimental data from? Top ↑
BIND, DIP, GRID, HPRD, IntAct, MINT, and PID.
From which databases does STRING extract curated data? Top ↑
Biocarta, BioCyc, GO, KEGG, and Reactome.
How do I extract purely experimental data? Top ↑
Uncheck all boxes, but the "Experiments" in the box "active interaction sources" under Data Settings tab.
I want to extract PPI for a given species, but only from experimental data and not from transferred from other species. Top ↑
Download the 'protein.links.full' from the download section of STRING. Using this file you can remove any combination of STRING channels using the above formula
How do I link to STRING networks? Top ↑
You can link to STRING in several ways.
To see the network neighborhood of a single protein the simplest way is to use our canonical links with STRING protein identifier attached at the end.
https://string-db.org/network/9606.ENSP00000234798
You can map your proteins to STRING proteins programmaticaly based on the 'alias' file found in the download section or by using our get string ids API.
Conviniently, you do not have to use our identifiers, as STRING recognizes a variety of protein synonyms including recommended protein names and their synonyms, UniProt/SwissProt accession, ENSEMBL identifiers etc. If species of a queried identifier is ambiguous STRING will try to resolve it, however it is recomended you add species name to our link in the following way:
https://string-db.org/network/homo_sapiens/TPSG1
You can find the list organisms and their names here
You can also link to a more specific STRING network of multiple proteins and with given flavour and neighborhood size. For example this is a 10 protein neighbohood network of TrpA and TrpB:
https://string-db.org/cgi/network.pl?identifiers=511145.b1260%0d511145.b1261
STRING understands a variety of other parameters which allows you to personalize your network.
| Parameter | value | Description |
|---|---|---|
| identifiers | protein name(s) | list of potein names seperated by '%0d' |
| add_color_nodes | 0 to 500 | adds color nodes to your network tarting from the most highly connected nodes (defailt 10) |
| add_white_nodes | 0 to 500 | adds white nodes to your network starting from the most highly connected nodes (default 0) |
| network_flavor | confidence,evidence,actions | the flavour of links between the proteins (default evidence) |
| required_score | 0 to 999 | the cut-off below which the links will not show (default 400) |
| channel[1-7] | on,off | select which evidences will contribute to the score (by default all are on) |
| caller_identity | your name | introduce yourself if your app is linking to STRING |
For example this call will show only the high confidence network neighborhood of TrpA and TrpGD:
https://string-db.org/cgi/network.pl?identifiers=511145.b1260%0d511145.b1263&add_white_nodes=10&add_color_nodes=0&required_score=950
If you don't want to link to STRING but instead just have an image of STRING network embedded in your website use our image API and just insert the call into the HTML image tag.
It is stated that STRING is locus-based and only a single translated protein per locus is stored. What does this mean? Top ↑
STRING uses one protein per gene. If there is more than one isoform per gene, we usually select the longest isoform, unless we have information that suggest that other isoform regarded as cannonical (e.g., proteins in the CCDS database).
Does STRING contain any Gene Ontology information? We see that there is a table called funcats. What type of information does this contain? Top ↑
The "funcats" contain the functional categories as defined for the COG database. We import the GO complexes and use these for inferring interaction. GO terms themselves are projected for future version.
Is there any phenotype information contained in STRING? More specifically, is there any field that specifies a phenotype or disease and links it to protein networks? Top ↑
Not directly, but by searching for "wing" in Drosophila will return genes that have been annotated/described as such, each of which is associated with a network.
Does the database give a PubMed Reference ID for each interaction? Top ↑
Interactions that have only predicted evidence do not have an PMID. Text-mining evidence may also stem from other sources, such as OMIM. Apart from the above, interactions come with at least one pubmed reference id. Some cases have several different and others have the same pmid (e.g., for external repositories, the interaction have the pmid of the publication of the database).
Are there different types of sets besides protein networks and pathways? What is the difference between a "set" and a "collection"? Top ↑
The different types of sets are networks, pathways, complexes, and PDB structures with more than one protein. The "sets_items" are members in the evidence sets. An interaction exists if two lines have the same set_id. The "sets" contain information of the set_ids, for example, from which "collection" they originate from. The "collections" are the different resources of data from which STRING imports data (for the channels 'experiments' and 'databases').
How do I access STRING using GI numbers. If it does, could you use 90 kD heat shock protein (GI:306891) as an example to let me know what should I type in protein name using NCBI GI number. Top ↑
The GI accession numbers are to track sequence histories of GenBank. STRING does use these number nor does it keep track of them, mainly because STRING is locus based. Also, STRING imports its sequences from Ensemble and RefSeq. If you need to cross reference to a particular entry in STRING from a GenBank record, you use the accession id of the GenBank nucleotide record. For example, 90kDa heat shock protein in human, will be M16660, which will give you this network.
Is there a legend or key for the different colored lines? (Is there a specific difference for each color?) (Is there a key for the colored lines in the evidence view?) Top ↑
Yes. You can always find the legend for your view under the "Legend" tab below the network.
I assume the arrows mean activation and the red perpendicular lines mean repression, but what to the circles at the end of the line represent? Top ↑
If we know a directionality of the action is indicated by the symbol at the end of the edge next to the protein that is acted upon. Down-Regulation is a red bar and up-regulation is a green arrow, as you say. Yellow circle describes that we know the directionality of the interaction e.g. ("A" acts upon "B"), but we do not know the result of the interaction (e.g., if it is up- or down-regulated).
At each node, there are icons inside the protein spheres. Is there a key for these icons? Do the icons represent the different protein functions (DNA binding, enzyme, etc.) Top ↑
The icons do not have any particular meaning other than that there is a structure associated with them. This can be either a PDB entry for the protein itself or a close homolog. If no PDB entry exists we look if their structure available by homology modeling from swiss-model. A small bubble (without icon) means that there is no structural information available. You can disable these structure previews in the "View Settings" tab.
I want to download the data for a particular network that I have found while browsing the STRING web-interface Top ↑
You can download your network in the "Tables / Exports" tab below your network. You can chose to download your data in a number of formats. The simplest to use is probably "Text Summary (TXT - simple tab delimited flatfile)".
I need all the interactions for a particular organism. Top ↑
You can download all data from the download section. At the bottom of the page there is a box where you can choose the organism of your interest. For example you can write "human" or "dog" there. When you click update all the files will automatically contain only the information about the taxon of your choice. STRING will also append ncbi taxonomy identifier prefix to each file-name.
Alternatively you can download not filtered file e.g. "protein.links.txt.gz" and parse it manually using the NCBI taxonomy identifier of the organism of your interest. You can find out here if the organism you are looking for exists in STRING along with its taxonomy identifier. Assuming you are using unix based operating system (including macs) you can parse the file like this (9606 is tax id of human):
zgrep "^9606\." protein.links.txt.gz > human.links.tsv
How to extract high confidence (>0.7) interactions from information on "combined score" in "protein.links.txt.gz" Top ↑
Here you can simply use awk to condition on the third column that contains the combined_score. Note that the scores are multiplied by 1000 to make them integers. I also assume that you only want evidence from human. Try the following:
zgrep "^9606\." protein.links.txt.gz | awk '($3 > 700) {print}'
How to retrieve only the direct evidence in human, not transferred. Top ↑
You need the file: "protein.links.full.txt.gz", from which you can retrieve the columns like above and write it to a file.
zgrep ^"9606\." protein.links.full.txt.gz | awk '($16 > 700) { print $1, $2, $3, $5, $6, $7, $8, $10, $12, $14, $16 }' > PPI_700_human.txt
The first and the second columns contains the STRING external identifiers. The last column contains the integrated scores including the homology transferred evidence.
In the file: "protein.links.txt" are the scores multiplied by 1000? Top ↑
Yes, the scores are multiplies by a factor 1000 (and truncated). 872 in the file means a STRING score of 0.872
Are the colors assigned to nodes significant? Top ↑
There colored nodes are your input (in case multiple-protein input) or first shell of interactors (in case of single-protein input). Grey nodes are proteins connected to your input or 2nd shell of interactors for multiple- and single input respectively. There is no particular meaning of the node color iteslef. They are used as a visual aid to identify which node goes with which description in list of input below the network ("Legend" tab) and in the evidence viewers.
What are the 1st and 2nd shell interactors? Top ↑
The 1st shell iteractors are the proteins directly associated with your input protein(s). 2nd shell of iteractors are the proteins associated with the proteins from the 1st shell or with your input protein(s). It can happen that a 2nd shell protein can be directly connected to your input protein(s), but it will usually have a weaker association and therefore it would not show up among the specified number of the 1st shell iteractors. You can recognize which shell the protein belongs to by looking at the color of the bubble, as 2nd shell proteins are always grey.
Why are some nodes smaller and some nodes bigger? Top ↑
The different size of the node only reflects that there is structural information associated with the protein. (i.e., it is larger to fit the thumbnail picture). You can disable the previews in the "View Settings" tab which will render the bubbles in the same size.
How do I map my proteins to STRING identifiers? Top ↑
You can use the file of 'protein.aliases.txt' available from the download page This file has four columns: species_ncbi_taxon_id, protein_id, alias, source. To figure out which is the string identifier for trpA in E. coli K12, you can do something like this in your terminal:
zgrep ^511145 protein.aliases.txt.gz | grep trpB
which would return:
511145 b1261 trpB BLAST_UniProt_GN RefSeq
from this you can get the string name by concatenating the two first column with a period (511145.b1261)
Is there an automatic way of mapping proteins to STRING? I need mappings for more three thousand proteins. Top ↑
A convenient way of mapping your proteins to STRING entries is to use the STRING'S API As an example, for a single protein, the alias can be retrieved by:
https://string-db.org/api/tsv/get_string_ids?identifiers=trpa&species=511145
Alternatively, instead of making on call per protein you can try to all the identifiers for a list of protein (separated by carriage return character '%0d'):
https://string-db.org/api/tsv/get_string_ids?identifiers=trpa%0dtrpb&species=511145
In such cases you may have a problems with the length limit of the URL, but this can be circumvented by sending the request as a HTTP POST request. For example using cURL:
curl -d "identifiers=trpA%0DtrpC%0DtrpB%0DtrpD\&species=511145" https://string-db.org/api/tsv/get_string_ids
The protein interactions from the STRING website via web API calls. What do the score columns mean (for example, nscore, fscore, tscore, etc)? Top ↑
Here is a summary.
- nscore - neighborhood score, (computed from the inter-gene nucleotide count).
- fscore - fusion score (derived from fused proteins in other species).
- pscore - cooccurence score of the phyletic profile (derived from similar absence/presence patterns of genes).
- hscore - homology score, the degree of homology of the interactors (normally not reported in STRING).
- ascore - coexpression score (derived from similar pattern of mRNA expression measured by DNA arrays and similar technologies).
- escore - experimental score (derived from experimental data, such as, affinity chromatography).
- dscore - database score (derived from curated data of various databases).
- tscore - textmining score (derived from the co-occurrence of gene/protein names in abstracts).
How do I select a reasonable score cut-off value for my analysis? Top ↑
You can use the score cut-off to limit the number of interactions to those that have higher confidence and are more likely to be true positives. Setting the cutoff lower, will increase coverage but also the fraction of false positives. You have to choose some arbitrary number based on the number of interactions you need for you analysis.
How do I import several interactions from STRING into Cytoscape. Top ↑
Cytoscape supports "tab separated values" file format. Download the "protein.links file" (from STRING download page), extract the interactions for you want (use grep or copy-paste), and load the processed file into cytoscape.
Limitation on the number of proteins? Top ↑
The web interface is not designed to handle large number of proteins and it is often difficult to visually interpret networks of large number of nodes. In such cases, it is better to process your data using the download files
Problems to download large files? Top ↑
To download files it is convenient to work in a terminal window. For example, the program "curl" with the option "-C -" is useful for downloading large files if you are on a shaky internet connection.
The downloaded file is really large. Which text editor should I use to view it? Top ↑
It is better not to open the file at all and extract the information from the file. On unix based systems (linux, mac) the safest way to peak and browse large files is to use cat/zcat (the latter is used with gzipped files) piped into less command.
zcat protein.links.v10.txt.gz | less -S
What is the Website Improvement Campaign? Top ↑
In order to further enhance the user experience on the STRING website, we occasionally run a 24-hour period of user activity tracking (about once every two months). This helps us to identify where our users get stuck, which parts of the website are preferentially visited, and which other parts might need improvements. During those periods, we track (anonymously!) all keyboard and mouse inputs on our website. The data are kept strictly confidential, and we only use them to analyze and improve the usability of our database. You will never receive any advertisements or other feedbacks resulting from this.
If you do not agree with this tracking, you can click the "deny" button in the header warning on top of the page, and the tracking will be turned off (this then applies also to future campaigns). If you want to change your mind, but the header warning is no longer visible, you can make STRING forget your previous choice by deleting the STRING "cookie" in your browser - then, the warning should re-appear after you've closed and re-opened the browser (see here to learn about cookies and how to delete them).
In general, please do consider allowing us to track your input during these restricted time windows. It greatly enhances our ability to further improve the user-friendliness of the website, and to identify those parts that are most important to you.